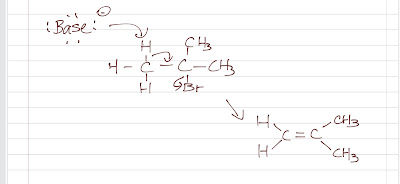
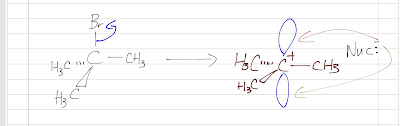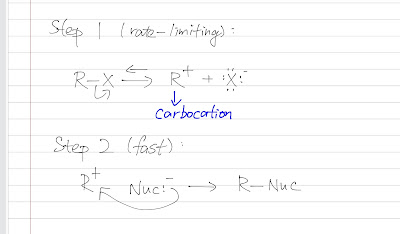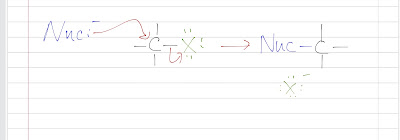EAS: Nitration

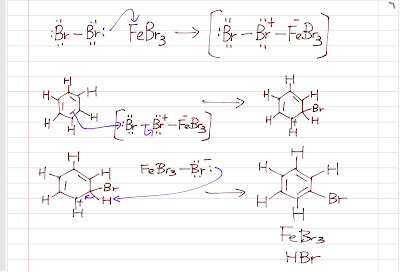
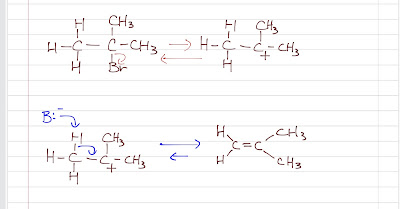
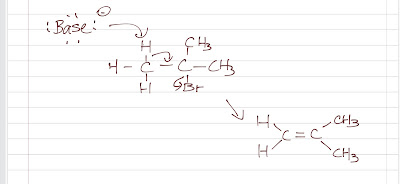
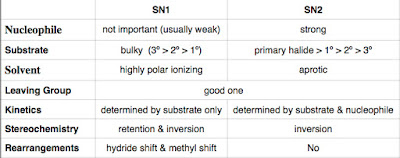
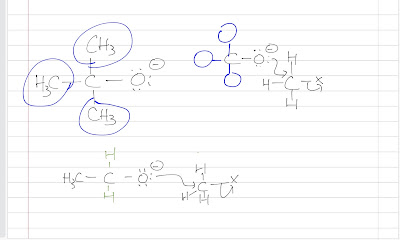
Nitration of benzene forms nitrobenzene . For it to take place, we need nitronium ion (strong electrophile ) to attack the benzene ring. We can do so by reaction nitric acid with an oxidizing agent. However, this will cause an explosion as the reaction is very exothermic. Therefore, we need a safer way, which is reacting nitric acid with sulfuric acid (catalyst, proton source). This way, we can form the electrophile , the nitronium ion. Sulfuric acid protonats the hydroxyl group to make it a better leaving group (water) to form nitronium ion. The reaction mechanism is as follows: After nitronium ion is formed, it is reacted with benzene. The electrophilic nitronium ion attacks the benzene ring. Deprotonation follows to from the conjugated system back. The nitro group of the product ( nitrobenzene ) can easily be reduced to amino group by treating with active metals (Zn, Fe, Sn) in dilute acid (HCl).